
La RMN arme majeure au service de la chimie et de la biologie structurale
Les états d'âme du spin dans un champ magnétique n'auraient pas de quoi passionner le chimiste, si, par exemple, tous les atomes d'hydrogène d'une molécule plongée dans un champ magnétique et soumis à un champ de radiofréquence se mettaient à résonner à l'unisson.
Heureusement il n'en est rien car notre atome d'hydrogène n'est pas isolé, en particulier il est lié à un autre atome (carbone, hétéroatome) par une liaison chimique constituée d'un doublet électronique.
Et là le phénomène se complique (encore !) et l'oreille du chimiste se dresse !
Ces électrons vont en effet produire en retour un champ magnétique secondaire qui va moduler le champ perçu par chaque noyau d'hydrogène. C'est le phénomène de blindage (ou de déblindage) qui fait que la résonance de chaque noyau est fonction de son environnement.
La différence, que l'on appelle déplacement chimique et que l'on mesure par rapport à une référence, est très petite, de l'ordre de 0,001% du champ appliqué.
Il y a donc une relation entre déplacement chimique et situation topologique de l'hydrogène dans la molécule. On comprend donc l'intérêt de la RMN pour le chimiste organicien qui veut établir une structure, sachant que l'hydrogène et le carbone sont les atomes les plus présents dans une molécule.
Avec un spectre du proton et du carbone (isotope 13) et toutes les techniques de découplage homo ou hétéronucléaires qui se sont développées (voir RMN 2D), le chercheur dispose d'armes d'identifications massives, qui font que même des structures complexes de macromolécules ont du mal à lui résister !
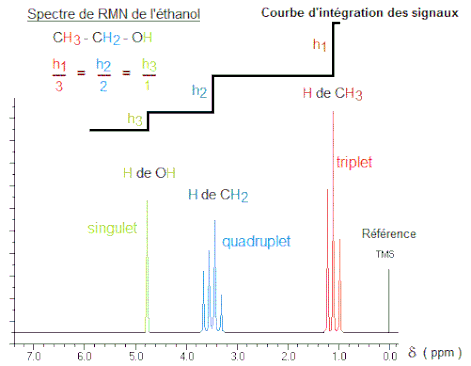
Il y a 40 ans ledit chercheur s'esbaudissait quand il découvrait le spectre d'une molécule basique comme l'éthanol.
On voit sur ce spectre trois groupes de signaux, correspondant aux trois types de protons non magnétiquement équivalents dans la structure.
On note que ces signaux présentent des multiplicités différentes qui sont dues au phénomène de couplage.
Ce couplage résulte de l'interaction de différents états de spin à travers les liaisons chimiques d'une molécule.
Encore une complexité précieuse qui indique (en première analyse) le nombre de protons présents sur le carbone vicinal. C'est la règle du n+1.
On voit que le signal de résonance des protons du méthyle (tous les 3 évidemment magnétiquement équivalents) est un triplet, ce qui correspond à un couplage avec les deux protons du carbone voisin (2+1).
Réciproquement le signal du méthylène est un quadruplet (couplage avec les 3 spins du méthyle). On observe que ce signal est plus déblindé que le précédent, ce qui traduit sa plus grande proximité avec l'oxygène, un élément très électronégatif.
On n'observe pas ici le couplage du proton du groupe hydroxyle, qui est donc un singulet.
Enfin, l'on sait que l'aire d'un signal est proportionnelle au nombre de protons qui lui correspond, ce qui permet de tracer une courbe d'intégration des signaux... et d'avoir encore un précieux renseignement en vue d'établir une structure.
La RMN à deux dimensions
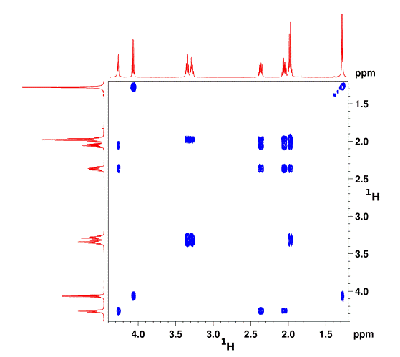
Aujourd'hui, avec les nouvelles techniques, on peut s'attaquer à des structures beaucoup plus complexes, d'intérêt biologique.
Outre l'analyse spectrale classique, avec les attributions des signaux, on peut réaliser des expériences à deux dimensions.
La RMN à deux dimensions a été proposée par J. JEENER (qui ne l'a pas publiée !) en 1971 ; il s'agit au départ d'utiliser des impulsions répétitives traitées par des transformées de Fourier multiples.
C'est R. Ernst de l'ETH Zurich qui est le père de la RMN impulsionnelle. Depuis, la technique a connue un essor formidable comme en témoignent les nombreuses applications en solution ou à l’état solide.
De très nombreuses séquences d'impulsions sont utilisées (COSY, NOESY, ROESY, COLOC...), mais le principe reste le même. Dans tous les cas elle permet d'établir des relations de proximité, dans l'espace ou à travers les liaisons, entre des noyaux actifs en RMN.
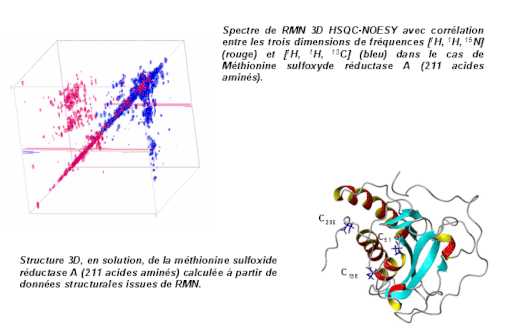
Ces corrélations peuvent être homonucléaires (proton-proton comme ci-dessus) ou hétéronucléaires (proton-azote15 comme ci-dessous).

Au spectre habituel se substitue donc une carte en 2D, sur laquelle apparaissent les résonances de chaque noyau, mais aussi les
interactions par paire de ces noyaux (corrélations), ce qui va permettre de connaître l'enchaînement chimique des atomes dans la molécule et même d'en apprécier la distance.
L'ensemble des données géométriques (distances inter-atomiques, angles dièdres...), collationnées, puis traitées par un calculateur, peuvent permettre de proposer, après itérations successives, une structure 3D.
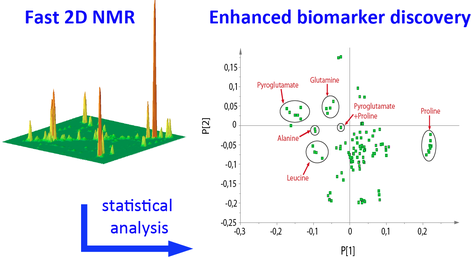
Les applications de la RMN 2D se multiplient et se diversifient.
Elle va par exemple permettre d'éviter le stade de la chromatographie dans l'analyse des mélanges.
A Gif-sur-Yvette, des mélanges chimiques et biochimiques très complexes, d’origines variées : des biofluides, des extraits bioactifs de plantes et d’organismes naturels... sont analysés par RMN 2D avec des outils de traitement de données.
C'est une technique très prometteuse pour des applications en métabolomique (science très récente qui étudie l'ensemble des métabolites d'origines diverses).
Au-delà de ces informations sur la structure tridimensionnelle des molécules, la RMN 2D est une méthode unique pour la caractérisation de la dynamique, à la fois globale et locale, des molécules sur des gammes de temps allant de la picoseconde à plusieurs mois.
En biologie structurale, où il s'agit de caractériser et de comprendre les mécanismes de reconnaissance moléculaires à l’échelle de d’atome, la RMN va permettre d'identifier les sites de contacts intermoléculaires, d'analyser les propriétés dynamiques des macromolécules et des interfaces, d'étudier le repliement des macromolécules, de mettre en évidence les interactions entre récepteurs et des cibles thérapeutiques potentielles... (voir les travaux du Laboratoire de Chimie et de Biologie Structurales de l’ICSN à Gif sur Yvette).
La RMN du solide
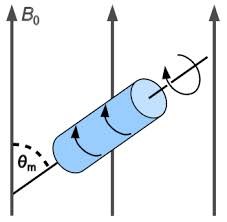
Les expériences que je viens de décrire concernent des molécules dissoutes dans un solvant, c'est la RMN en solution. L'échantillon à analyser est dans un tube parfaitement cylindrique, qui, après son introduction dans la sonde, va être mis en rotation très rapide de façon à assurer une parfaite homogénéité du champ magnétique perçu par l'ensemble des spins. En effet, cet échantillon contient un très grand nombre de molécules, il faut donc obtenir un champ B0 le plus homogène possible dans tout le volume qu'il occupe, de façon à ce que deux protons analogues sur deux molécules différentes produisent des signaux identiques qui pourront se cumuler.
La contrainte d'homogénéité du champ statique est capitale.
Avec la 2D, la RMN en solution est donc capable d'établir des structures
tridimensionnelles à l'instar des études
par RX qui nécessitaient la cristallisation des molécules à étudier, ce qui excluaient nombre de macromolécules.
Cependant, la question qui s'est rapidement posée aux rmnistes et aux (bio)chimistes était de savoir comment il serait possible de réaliser des expériences de RMN sur des composés solides.
L'obstacle était de taille.
En effet, dans les solides, il existe des interactions anisotropes supplémentaires (qui s'annulent en solution grâce au mouvement brownien) : le couplage dipolaire (interaction directe entre les dipôles magnétiques, à travers l’espace) et l’anisotropie de déplacement chimique. Ces interactions modifient les niveaux d'énergie des spins nucléaires (et donc la fréquence du signal de résonance) et provoquent un fort élargissement du signal.
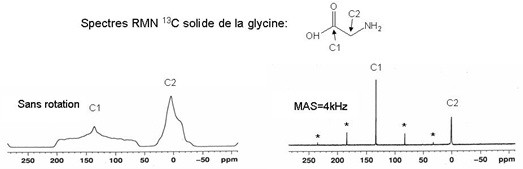
La RMN du solide peut présenter un intérêt pour le chimiste du vivant dans l'étude de grosses molécules amorphes et peu solubles, de polymères ou du suivi des synthèses dur support.
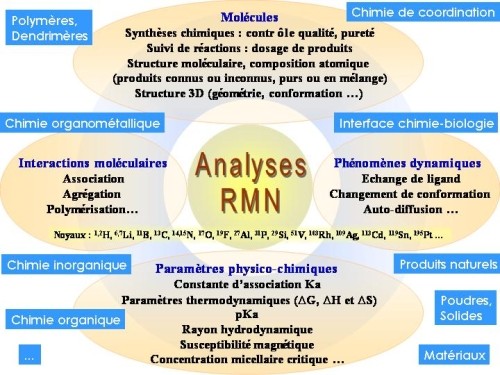
La diapositive ci-dessous résume le potentiel extraordinaire de la RMN à l'usage du chimiste, du biologiste structuraliste et de tous ceux qui travaillent à l'interface chimie-biologie.
La RMN au centre de toutes les investigations concernant le vivant, l'approche "omique"

Initialement utilisée pour la simple détermination de structure des molécules, l'analyse RMN est maintenant devenue incontournable dans toutes les problématiques concernant le vivant et en particulier dans tous les phénomènes dynamiques complexes qui font aujourd'hui l'objet de recherches intensives.
La mise au point de spectromètres à très haut champ, combinée aux techniques multidimensionnelles et aux progrès de l’informatique, font de la RMN un outil de choix dans la détermination de la structure tridimensionnelle d’une protéine et donc dans l'étude du protéome humain.
La caractérisation de toutes les protéines d’un protéome va en effet permettre une meilleure compréhension de la relation structure/fonction des protéines d’un organisme. De nombreux projets de protéomique structurale sont en cours en Europe, aux USA et au Japon.
La RMN est aussi élément central des plate-formes métabolomiques qui se mettent en place, au côté de la spectrométrie de masse couplée, pour l'étude de systèmes complexes pouvant permettre d'identifier les fluctuations physiologiques et pathologiques du métabolome, conduisant ainsi à la détection de marqueurs (pronostics, précoces ou descriptifs).
La résonance magnétique nucléaire est donc un des piliers de l'approche omique : science qui incorporent diverses disciplines telles que la génomique, la protéomique, la transcriptomique et la métabolomique; fournissant avec la spectrométrie de masse un grand nombre de données traitées par les méthodes de la bioinformatique.
Si la RMN est vraiment l'outil fondamental du chimiste, celui-ci dispose aujourd'hui d'une palette impressionnante de méthodes spectroscopiques. Au fil de ce récit j'en évoque quelques unes comme la spectroscopie de masse, les RX...
Molécules : il faut les voir pour le croire !
A l'orée des années 70, au moment où je faisais mes premiers pas dans un laboratoire de recherche, les méthodes spectroscopiques modernes (résonance magnétique nucléaire et spectrométrie de masse notamment) commençaient à être utilisées de façon routinière. C'était évidement un apport considérable pour le suivi des réactions chimiques et nous étions plein d'admiration pour nos prédécesseurs qui avaient su faire de la bonne chimie sans ces outils.
Cependant il s'agissait alors de déterminer la structure de petites molécules.
Au début des années 80, la chimie organique est devenue la chimie du vivant et le chimiste a commencé à observer, puis à manipuler, de grosses molécules, des macromolécules, puis des édifices supramoléculaires de taille nanométrique.
Heureusement, grâce aux mathématiciens, physiciens et surtout informaticiens, la spectroscopie faisait également sa révolution. Adieu spectres d'antan, bienvenue à l'imagerie.
Dès lors la course aux armements ne cessera plus, d'autant que, depuis le mitan des années 2000, le chimiste du vivant se trouve impliqué dans la formidable aventure de la biologie de synthèse, dont le but est de comprendre les systèmes biologiques complexes dans leur globalité et dès lors de concevoir de façon rationnelle de nouveaux organismes ayant des fonctions biologiques.
On touche là à l'essence même du vivant avec toute la complexité et la discrétion qui s'y rattache. Il est clair que pour percer ce mystère, le scientifique doit être solidement équipé.

Détermination de structures macromoléculaires par diffraction des rayons X
Tomodensitométrie de la molécule : le scanner moléculaire

Cette nouvelle technique, présentée par des chercheurs californiens, est qualifiée de révolutionnaire par de nombreux chimistes, notamment par ceux qui sont impliqués dans la "drug discovery" (recherche de médicaments).
C'est en fait une technique de diffraction des rayons X où le cristal pivote. L'évolution du diagramme de diffraction fournit une image qui ressemble à celle d'une tomodensitométrie moléculaire.
Elle permet d'obtenir des structures à partir de cristaux d'un milliardième de la taille de ceux nécessaires à la cristallographie à rayons X.
Cette équipe annonce même avoir obtenu des structures à partir de mélanges de composés et de matériaux qui n’avaient jamais été cristallisés formellement.